Research
Our group analyses molecular mechanisms that control tumor development and progression. We are particularly interested in the regulation of the oncogenic Wnt signal transduction pathway.
Molecular oncology of Wnt signaling
The Wnt signaling pathway controls the stability of β-catenin and thereby regulates various processes during embryonic development and can lead to cancer. Wnt are secreted glycoproteins, which induce the accumulation of β-catenin in cytoplasm and nucleus by binding to frizzled and LRP receptors. β-Catenin interacts with TCF transcription factors and activates target genes. The destruction of b-catenin is induced by phosphorylation in a multi-protein complex consisting of the scaffold components axin or conductin, the serine/threonine kinase GSK3β, and the tumor suppressor APC (Adenomatous Polyposis Coli). The Wnt signal inhibits phosphorylation of β-catenin and thereby leads to its stabilization. In colorectal tumors, mutations of APC or of the serine/threonine phosphorylation sites of β-catenin lead to stabilization of β-catenin and trigger constitutive signaling to the nucleus. Such β-catenin mutations are also found in a multitude of other tumor types suggesting that aberrant activation of Wnt signaling is a key mechanism of oncogenic transformation. We are analyzing the molecular roles of central components of the pathway, which are mostly involved in β-catenin degradation. Among these are Amer1, Axin, Conductin as well as the phosphatase PGAM5 that all modulate β-catenin phosphorylation.
Amer proteins
Amer1 (APC membrane recruitment1) also called WTX is the best characterized member of the Amer protein family, which also includes Amer2 and Amer3. Amer1 interacts with APC and can recruit it to the plasma membrane, thereby acting as a negative regulator of Wnt signaling. The Amer1 gene is mutated in up to 30% of Wilms tumors, but also in 7 – 12% of colorectal carcinomas. Moreover, Amer1 mutations underlie the inherited disease OSCS, which is characterized by bone malformations and defects in other organs. In order to determine the consequences of Amer1 mutation for tumorigenesis in vivo, we conditionally knocked-out the Amer1 gene specifically in gut epithelium by crossing floxed Amer1 mice with villin-Cre mice. Efficient depletion of Amer1 in intestinal epithelial cells was verified by genetic means and RT-PCR analysis. Detailed analysis of histological sections revealed no alterations of epithelial cell proliferation and differentiation after Amer1 loss. There was also no signs of tumorigenesis following Amer1 depletion even after more than nine months of inspection. This indicates that Amer1 does not act as a tumor suppressor in mice in the absence of other oncogenic mutations. Since Amer1 mutations co-occur with APC mutations in most of the colorectal cancer cases, and APC is the central tumor suppressor and gatekeeper of these tumors, we crossed conditional Amer1 k.o. mice with APCmin mice in which the APC gene is mutated. APCmin develop tumors (polyps) mainly in the small intestine, but also at a lower rate in the colon a few months after birth. An initial analysis after three months did not show an impact of Amer1 depletion on polyp number and size in the APCmin mice.
Role of Axin/Conductin as negativeWnt regulators
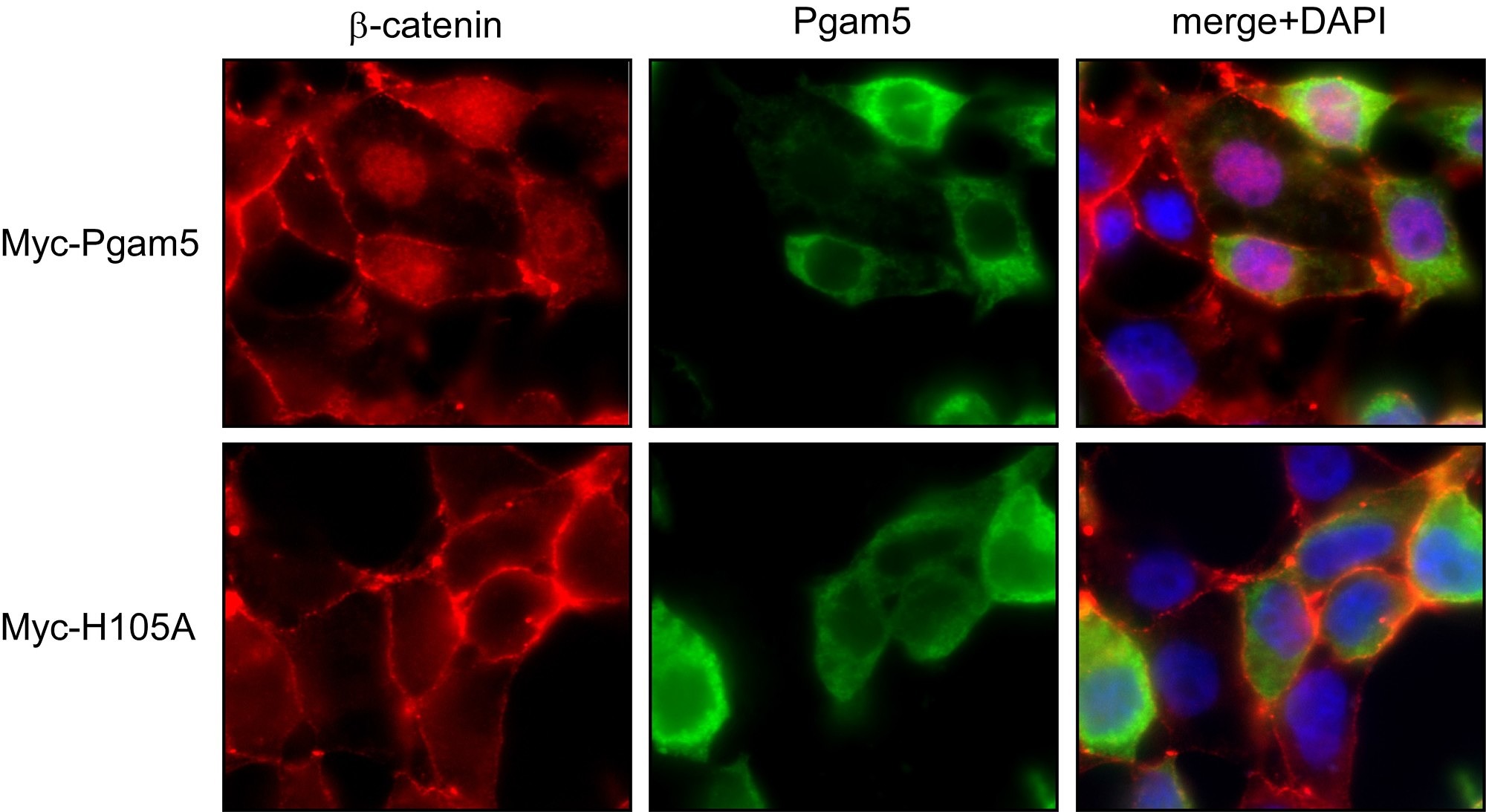
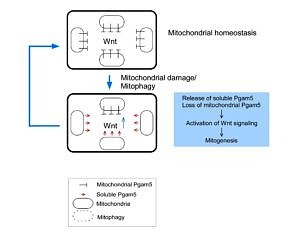
Axin and conductin (also known as axin2) are structurally related inhibitors of Wnt/β-catenin signaling that promote degradation of β-catenin. Whereas axin is constitutively expressed, conductin is a Wnt target gene implicated in Wnt negative-feedback regulation. By proteome analysis we could demonstrate an interaction of axin with the mitochondrial phosphatase PGAM5. We found that knockdown of PGAM5 activates the Wnt pathway. However, we also found that PGAM5 is cleaved and released to the cytoplasm after damage of the mitochondrial membrane potential. Cytoplasmic PGAM5 interacts with axin, and the resulting complex induces dephosphorylation of β-catenin leading to its stabilization and activation of β-catenin-dependant transcription. It was previously shown by others that Wnt stimulation leads to mitochondrial biogenesis, pointing to a role of the PGAM5-Axin-β-catenin axis in mitochondrial homeostasis. Indeed, overexpression of cytoplasmic PGAM5 sufficed to increase mitochondrial numbers by a factor of approximately 1.5. On the basis of our results we propose the following model: mitochondrial damage associated with a loss of membrane potential leads to the release of cytoplasmic PGAM5 which stimulates Wnt signaling resulting in formation of new mitochondria that compensate for the initial loss of mitochondria.
|
|
|